I sistemi biologici nelle simulazioni coarse grained multi-scala
Studiare un sistema biologico con un modello matematico può essere molto vantaggioso, ma a volte i dettagli da tenere in considerazione sono tantissimi. Per questo può essere utile analizzare il sistema con due scale diverse, ad alta e a bassa risoluzione.
RICERCANDO ALL’ESTERO – Da diversi anni l’uso dei modelli matematici è entrato a far parte a tutti gli effetti dello studio dei sistemi biologici complessi, portando a un risparmio di tempo e risorse computazionali. Tuttavia, la simulazione di sistemi come quelli di materia soffice spesso comprende parametri difficili da conciliare, come le grandi dimensioni e il numero di dettagli fisici e chimici necessari per descrivere in modo sufficientemente preciso un certo fenomeno.
Per risolvere questi problemi sono stati sviluppati approcci multi-scala, in grado di studiare contemporaneamente diverse regioni di uno stesso sistema a diversi livelli di risoluzione.
Cosa si intende per sistemi di materia soffice?
Il termine comprende uno spettro molto ampio di sistemi: secondo una definizione estremamente lasca ci si riferisce a tutto ciò che è soffice al tatto umano, dai liquidi alla materia biologica, ai polimeri e perfino al cibo. In modo un po’ più formale, sono quei sistemi con una caratteristica scala di energia, prossima a quella corrispondente alla temperatura ambiente in fisica.
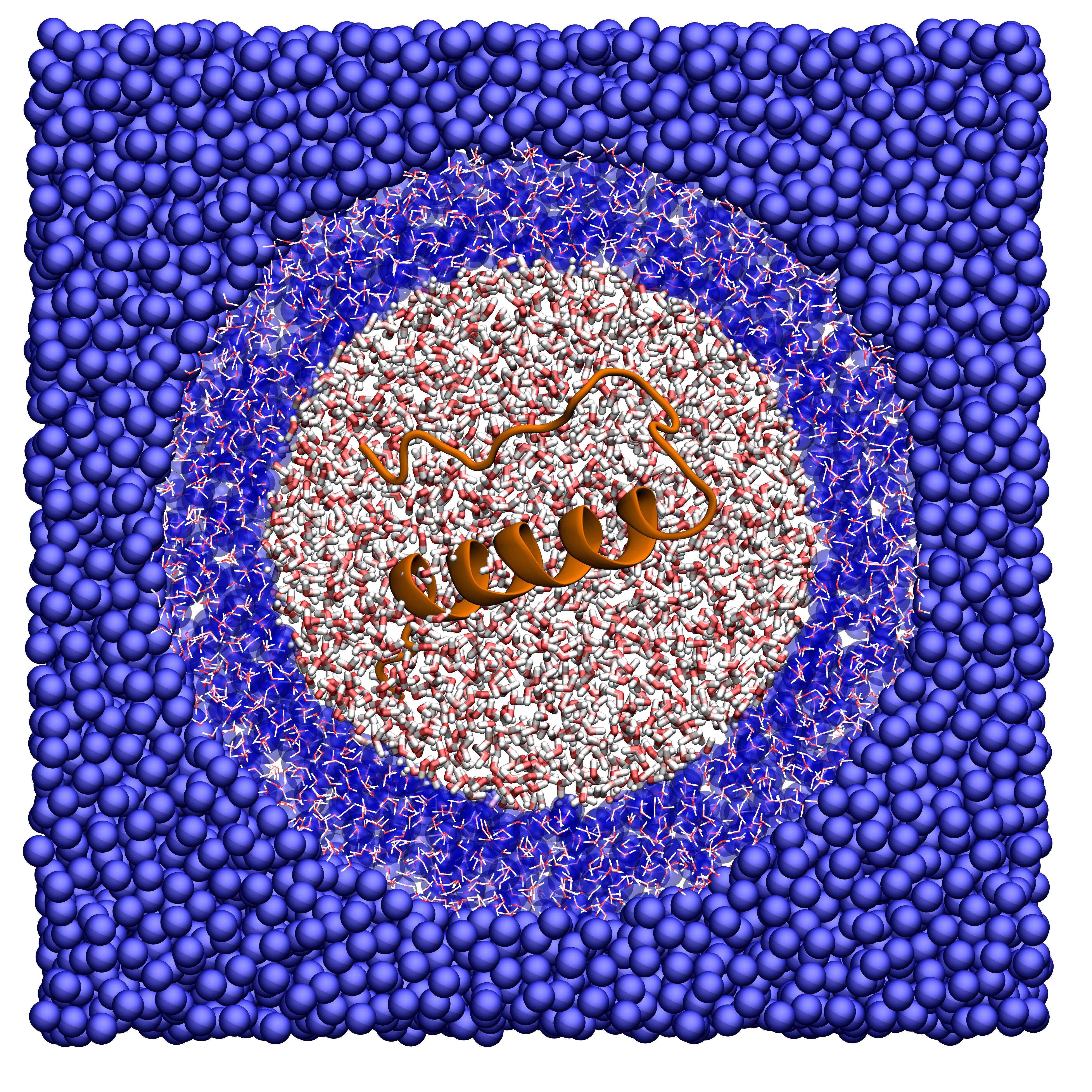
Il mio lavoro consiste nello studiare la materia soffice attraverso modelli coarse grained: si tratta di rappresentazioni in cui la risoluzione, quindi il dettaglio con cui descriviamo un certo sistema, non è di tipo atomistico ma per unità fondamentali. Con questi modelli, per esempio, le molecole d’acqua non vengono descritte con due atomi di idrogeno e uno di ossigeno, ciascuno con le sue interazioni, ma con un solo punto materiale che interagisce in modo efficace con altri punti materiali.
Così facendo si semplifica molto la descrizione del sistema ma, se le unità fondamentali vengono scelte in modo accurato, la rappresentazione rimane quantitativamente fedele e abbastanza precisa. Nel processo di costruzione del modello, quindi, ben prima della simulazione, è fondamentale analizzare tutti i dettagli del problema in modo da capire quali sono gli ingredienti essenziali per la fisica del sistema e quali le caratteristiche molto specifiche del fenomeno in oggetto.
Quali strategie si usano nella costruzione del modello?
Dipende dal tipo di informazioni che si vogliono ottenere, se più quantitative rispetto a ciò che succede nel sistema o più qualitative su ciò che guida il fenomeno.
Di questo secondo caso fanno parte i modelli che descrivono in modo molto semplificato le proteine che formano le fibrille amiloidi coinvolte nello sviluppo di malattie neurodegenerative come l’Alzheimer. Le proteine vengono rappresentate come rigidi parallelepipedi, descritti a livello puramente geometrico, che si attaccano gli uni agli altri quando si trovano abbastanza vicini tra loro. Un modello del genere quantitativamente non ha nessuna relazione con il sistema reale ma contiene gli ingredienti giusti per descrivere a livello qualitativo il processo di formazione delle fibrille amiloidi.
La strategia di costruzione più quantitativa, invece, parte da una descrizione del sistema a risoluzione atomistica, identifica una quantità caratteristica da usare come riferimento (per esempio le proprietà termodinamiche), e usa un procedimento algoritmico per costruire il modello coarse grained a bassa risoluzione. Non tutte le proprietà verranno riprodotte correttamente, ma quelle usate per parametrizzare il sistema saranno, per costruzione, conservate.
Esistono infine strategie ibride (o multi-scala), in cui all’interno della stessa simulazione si usano due modelli diversi a risoluzione diversa: uno coarse grained per descrivere il sistema in generale e uno atomistico per studiare in modo dettagliato una piccola parte di esso. Il vantaggio di questo approccio è il risparmio di risorse computazionali: i modelli coarse grained hanno meno centri di interazione (meno particelle), meno forze da calcolare, sono più veloci nelle simulazioni e quindi possono essere usati più a lungo e per sistemi più grandi. Nei casi in cui la loro bassa risoluzione non è sufficiente a caratterizzare in modo adeguato il fenomeno di interesse, si passa a una simulazione atomistica.
Che tipo di sistemi si possono studiare con i modelli coarse grained multi-scala?
Un tipico esempio è lo studio del sito catalitico di un enzima, in cui l’interazione con la molecola da legare dipende fortemente dalla disposizione degli atomi nello spazio. Il sistema in generale può essere descritto dal modello coarse grained, usando magari un unico sito di interazione per ciascun amminoacido e quindi riducendo il numero di particelle coinvolte; il modello atomistico poi simula quella piccola regione proteica che non può prescindere da una risoluzione accurata.
La cosa difficile è far sì che queste due parti del sistema (ad alta e bassa risoluzione) si parlino tra loro in modo amichevole, ovvero che la fisica rimanga corretta e la parte ad alta risoluzione non risenta negativamente di quella a bassa risoluzione.
Un altro ambito è lo studio dei liquidi: qui la complicazione sta nelle molecole del liquido che non sono immobili e possono entrare e uscire dalla zona ad alta risoluzione, per cui ogni volta bisogna cambiare al volo la loro descrizione.
Pensiamo all’assemblaggio dei capsidi virali, cioè del guscio dei virus: sarebbe impensabile fare una simulazione atomistica di un sistema così sofisticato con le risorse computazionali che abbiamo oggi. Nei modelli coarse grained le proteine virali vengono trattate come mattoncini lego di forma e interazioni particolari e, pur perdendo informazioni sulla sequenza, struttura e sulle interazioni atomistiche delle molecole coinvolte, riescono a riprodurre la formazione del capside completo e perfino a ottenere informazioni quantitative sul tempo impiegato.
Infine, da decenni si fa una fatica enorme a sviluppare nuovi farmaci, inventando molecole sempre nuove e verificando se e come funzionano: farlo attraverso saggi sperimentali porta via tantissimo tempo, mentre uno screening a livello computazionale permette di identificare i candidati più promettenti da testare in laboratorio in 10 o addirittura 100 volte meno tempo.
Quali sono le prospettive future del tuo lavoro?
Applicare questi modelli multi-scala a problemi specifici, in particolare a sistemi biologici e grandi biomolecole.
Ultimamente ci stiamo concentrando sullo studio di biopolimeri – cioè proteine e DNA – annodati: vogliamo capire quali sono gli ingredienti fondamentali che portano le proteine a seguire una sequenza di eventi ben definita, annodarsi e a completare la formazione della loro struttura tridimensionale. Nel modello che abbiamo creato, ciascun amminoacido è descritto da una sfera, le interazioni sono tutte dello stesso tipo e si considera solo il volume escluso (in modo che due amminoacidi non si possano sovrapporre); vengono rappresentate anche interazioni specifiche per ciascuna proteina, analoghe a quelle che determinano gli angoli tra triplette e quartetti di amminoacidi in sequenza.
 Nome: Raffaello Potestio
Nome: Raffaello Potestio
Età: 34 anni
Nato a: Roma
Vivo a: Wiesbaden (Germania)
Dottorato in: fisica statistica e biologica (Trieste)
Ricerca: Modellizzazione multi-scala di materia soffice
Istituto: Statistical Mechanics of Biomolecules, Max Planck Institute for Polymer Research (Mainz, Germania)
Interessi: politica, cinema, stare con mia figlia, leggere
Di Wiesbaden mi piace: la primavera e l’estate, passeggiare nei parchi; è delle poche città risparmiate dai bombardamenti, adoro il suo stile neoclassico e liberty
Di Wiesbaden non mi piace: è tutto troppo silenzioso
Pensiero: If it can work, it will
Leggi anche: Modelli computazionali per lo sviluppo di nuovi farmaci
Pubblicato con licenza Creative Commons Attribuzione-Non opere derivate 2.5 Italia.