Patologie prioniche: non solo mucca pazza
I prioni sono proteine "impazzite" responsabili di malattie neurodegenerative in umani e animali. La ricerca scientifica continua, con risvolti promettenti anche per Parkinson e Alzheimer.
I prioni sono il primo esempio riconosciuto di agente patogeno che non sia un virus, un batterio, un fungo o un parassita. Per i primi anni successivi alla loro scoperta, avvenuta negli anni ’60, l’ipotesi più accreditata era che fossero virus atipici, mentre oggi la maggior parte degli studiosi è concorde nel considerarli privi di acidi nucleici, ossia di informazione genetica. Cosa sono, allora? E perché sono così pericolosi?
Una questione di struttura
Le proteine, molecole fondamentali per la sopravvivenza di qualsiasi animale, essere umano compreso, sono caratterizzate da strutture tridimensionali molto complesse. Per giungere a queste architetture elaborate, fondamentali affinché le proteine svolgano correttamente la propria funzione, le catene di amminoacidi che le costituiscono subiscono un vero e proprio processo di ripiegamento. Non sempre, però, tutto va per il verso giusto: le proteine ottenute tramite un ripiegamento errato (chiamate in gergo misfolded) sono instabili e agiscono in maniera incontrollata con gli altri tessuti.
Tra queste proteine “impazzite” ci sono anche i prioni, derivati da una proteina chiamata PRNP, espressa a livello di membrana cellulare.
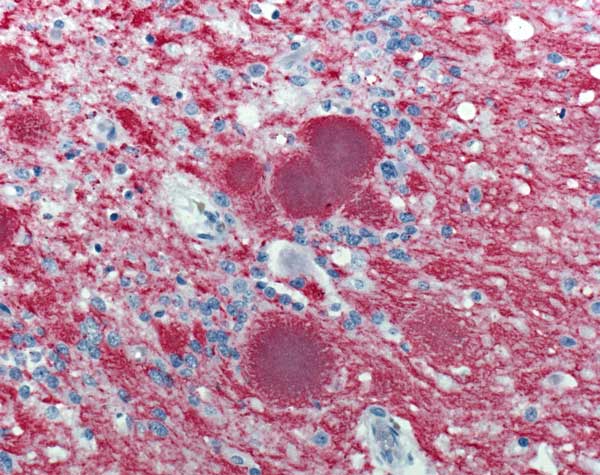
I prioni sono responsabili dello sviluppo di malattie neurodegenerative come la Creutzfeldt-Jakob Disease (CJD) negli esseri umani, l’Encefalopatia Spongiforme Bovina (conosciuta anche come BSE o con l’immagine colorita di “morbo della mucca pazza”) nei bovini, la Scrapie negli ovini e la Chronic Wasting Disease nei cervidi. In questi disturbi, le isoforme anomale di PRNP si aggregano in fibre altamente strutturate, chiamate amiloidi, che si accumulano in placche (simili a quelle osservate nella Malattia di Alzheimer).
A oggi, però – e questo è esemplificativo di quanto la comprensione di queste patologie sia ancora lungi dall’essere esaustiva – non è chiaro se siano queste placche a causare il danno e la successiva morte cellulare delle cellule nervose, o se invece rappresentino un effetto del processo patologico in atto.
Quale che sia il fattore scatenante, ciò che si osserva in maniera simile in queste patologie è la formazione di veri e propri buchi all’interno del tessuto nervoso, che conferiscono allo stesso una struttura spongiforme. La sostanziale disgregazione della normale citoarchitettura neurale si traduce quindi in demenza, atassia (incapacità di rimanere in equilibrio e di coordinare i movimenti), convulsioni e, nell’essere umano, disturbi del comportamento e della personalità.
Una rete per il monitoraggio
La via più comune attraverso la quale contrarre una patologia prionica è l’ingestione di tessuto infetto; a ciò è dovuto il clima di allarme (sfociato a volte in veri e propri momenti di psicosi collettiva) che ha dominato l’Europa a cavallo tra gli anni ’80 e ’90, quando carni provenienti da bovini infetti da BSE (che a loro volta erano stati contagiati a causa dell’ingestione di farine di origine animale) hanno causato un’impennata di casi di CJD nell’uomo.
Inoltre, i prioni si possono depositare nell’ambiente attraverso la decomposizione delle carcasse di animali morti, dove possono andare a legarsi ad argilla e altri materiali con cui poi l’essere umano può entrare in contatto. Un’altra via attraverso cui può avvenire il contagio è il contatto con urina, saliva e altri liquidi corporei da parte di un animale o di un individuo infetto. Un team di ricerca guidato da Stanley Prusiner, vincitore del premio Nobel per la Medicina nel 1997, ha ipotizzato che l’infezione possa trasmettersi anche attraverso i prioni presenti nel letame, largamente utilizzato in agricoltura.
A fronte di patologie incurabili, fatali, caratterizzate da una trasmissione così variegata, da un periodo di incubazione relativamente lungo e da una progressione sintomatologica molto veloce, è perciò importante istituire una rete di sorveglianza e monitoraggio per individuare tempestivamente casi sospetti. Proprio per questo agli inizi degli anni’90 è stato istituito l’European Creutzfeldt-Jakob Disease Surveillance Network (EuroCJD), che raccoglie professionisti coinvolti nella ricerca, nella diagnosi e nel trattamento di queste patologie.
“Tipicamente un medico, di medicina generale o un neurologo, individua un possibile caso di Creutzfeldt-Jakob Disease e ci contatta”. A spiegarlo è Gabriele Piconi, ricercatore del Centre for Clinical Brain Science (CCBS) di Edimburgo e membro dell’Unità di Sorveglianza scozzese di EuroCJD. “A quel punto noi possiamo intervenire a diversi livelli: a livello clinico, i nostri neurologi possono assistere il medico che ha segnalato il caso sospetto nella gestione del paziente e dei suoi familiari; parallelamente, a livello diagnostico, i tecnici di laboratorio possono eseguire le analisi di routine necessarie per identificare la presenza di una patologia riconducibile ai prioni”.
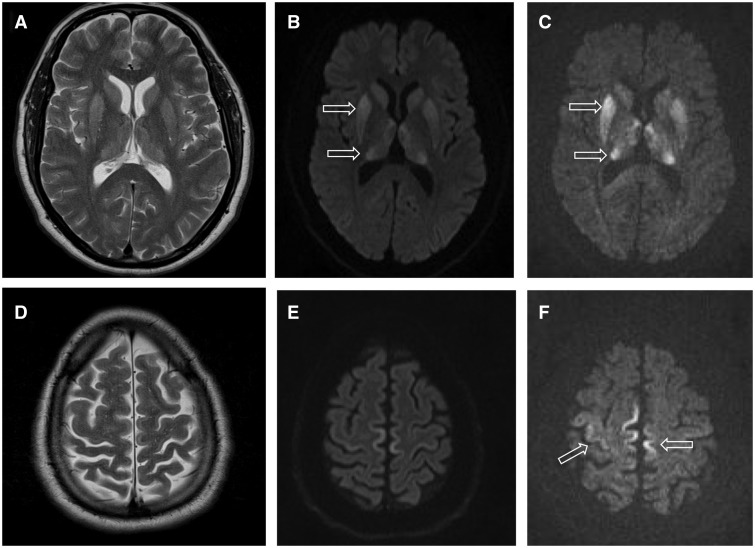
Tuttavia, per la CJD e per altre patologie prioniche, parlare di una vera e propria diagnosi non è possibile, fino a quando non è ormai troppo tardi. “In accordo con le linee guida dell’Organizzazione Mondiale della Sanità, che EuroCJD ha contribuito a stilare e aggiornare, a oggi la diagnosi definitiva si raggiunge solo post mortem, tramite un’accurata analisi istopatologica del tessuto cerebrale” spiega infatti il ricercatore.
I campioni così raccolti nel corso degli anni (che includono tessuti cerebrali, ma anche sangue, urine, tessuti linfoidali e liquido cerebrospinale) hanno contribuito a realizzare un corposo database, cruciale non solo per la profilassi legata a queste patologie ma anche per la ricerca volta ad identificarne cause e possibili trattamenti, la cosiddetta biobanca della CJD.
“La biobanca è fondamentale per lo studio di queste patologie, causate da patogeni che, a differenza di batteri o virus, non possono essere mantenuti e amplificati in vitro. È solo grazie alla disponibilità delle famiglie dei pazienti che abbiamo a disposizione dei prioni nella forma in cui essi si sviluppano dell’uomo”. Visto il carattere peculiare che queste patologie acquisiscono nelle differenti specie, è ovviamente preferibile alla ricerca condotta su modelli animali, che comunque esistono.
Studiare una malattia per capirne tante
Molti sforzi dell’odierna ricerca scientifica che gravita intorno alle patologie prioniche si basano sullo sviluppo di tecnologie di diagnosi più accurate: le più moderne, i cosiddetti cell-free conversion systems, sfruttano il meccanismo di riproduzione dei prioni. Tra questi metodi, il più promettente sembra l’RT-QuIC, un saggio di amplificazione del prione, in grado di rilevare la forma anomala della proteina a concentrazioni bassissime.
L’importanza di queste tecniche non è però relegata alle sole patologie prioniche, come spiega Piconi. “C’è una ricchissima letteratura scientifica [qui un esempio] che sostiene l’ipotesi di un modello simil-prionico di diffusione di proteine cruciali nello sviluppo di altre patologie neurodegenerative, come l’alfa-sinucleina nella Malattia di Parkinson e la proteina tau in quella di Alzheimer”.
Anche in questi casi, come per le patologie causate dai prioni, la diagnosi precoce e lo sviluppo di trattamenti terapeutici non invasivi rappresentano un traguardo importante, ambizioso ma non impossibile. È in questa direzione che si concentreranno gli sforzi di clinici e ricercatori nei prossimi anni.
Leggi anche: Uno sguardo alla malattia di Parkinson
Articolo pubblicato con licenza Creative Commons Attribuzione-Non opere derivate 2.5 Italia. ![]()



