Terapie personalizzate per la fibrosi cistica
La fibrosi cistica è una malattia complessa, con sintomi e gravità che variano da soggetto a soggetto. Anche la risposta dei pazienti ai farmaci è molto variabile e studiare a fondo tutti i fattori coinvolti è importante per mettere a punto terapie personalizzate efficaci.
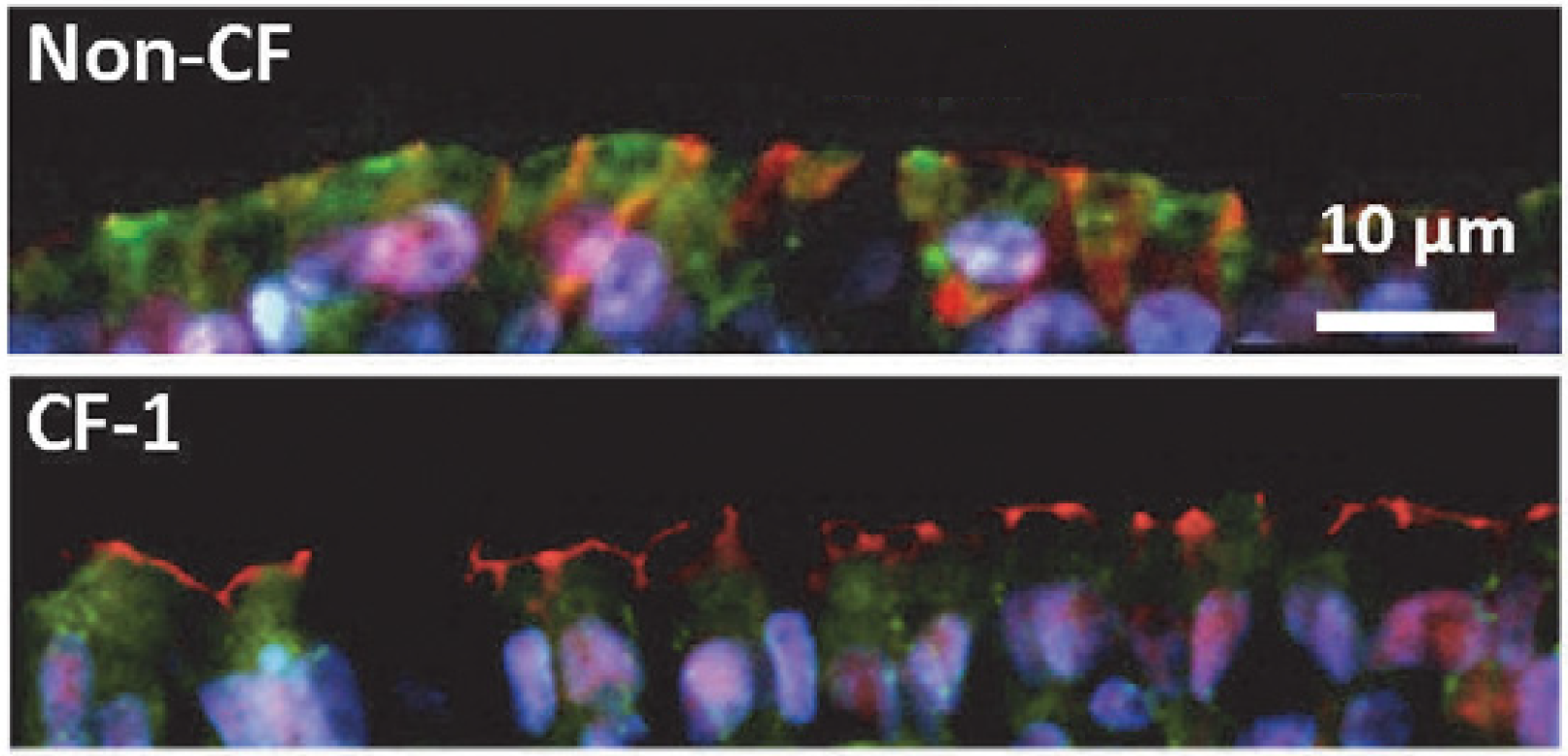
La fibrosi cistica è una malattia genetica grave dovuta a un difetto nella proteina CFTR (Cystic Fibrosis Transmembrane Conductance Regulator). CFTR è un canale del cloro coinvolto nella produzione di muco, sudore, saliva, lacrime ed enzimi digestivi: se non funziona correttamente, queste secrezioni diventano più dense e appiccicose, al punto da ostruire le vie respiratorie e le ghiandole. I sintomi più conosciuti sono a carico dei polmoni e del pancreas, anche se la fibrosi cistica è una malattia multiorgano che colpisce un po’ tutto il corpo.
Non esiste una cura per la fibrosi cistica ma grazie alle terapie in alcuni casi l’aspettativa di vita è salita a 42-50 anni.
Onofrio Laselva è ricercatore al Hospital for Sick Children di Toronto (Canada) e si occupa di individuare nuovi composti per ripristinare e potenziare la funzione di CFTR.
 Nome: Onofrio Laselva
Nome: Onofrio Laselva
Età: 30 anni
Nato a: Conversano (BA)
Vivo a: Toronto (Canada)
Dottorato in: fisiologia (Bari)
Ricerca: Medicina traslazionale per pazienti con fibrosi cistica
Istituto: Dipartimento di medicina molecolare, Hospital for Sick Children (Toronto, Canada)
Interessi: viaggiare, stare con gli amici
Di Toronto mi piace: è molto sicura per essere una metropoli, non è confusionaria, ha una bellissima natura
Di Toronto non mi piace: le persone sono un po’ fredde, non c’è un’integrazione completa tra le varie culture
Pensiero: Non c’è nessun ostacolo che ci impedisce di raggiungere quel che noi desideriamo, l’ostacolo è nella nostra mente!
Come viene valutata l’efficacia di potenziali nuovi farmaci per la fibrosi cistica?
Siamo in collaborazione con diverse case farmaceutiche che sintetizzano composti con una potenziale azione su CFTR. Per verificare quali sono effettivamente in grado di riportare l’equilibrio tra ioni e acqua nei tessuti, facciamo uno screening farmacologico su cellule in vitro. In particolare testiamo due tipi di molecole, correttori e potenziatori: i correttori portano a un ripristino di CFTR sulla membrana apicale delle cellule mentre i potenziatori favoriscono l’apertura del canale CFTR mutato (difettoso).
CFTR è una proteina composta da diversi domini, MSD1 e MSD2 attraversano la membrana delle cellule, NBD1 e NBD2 interni al citoplasma legano i nucleotidi e R ha funzioni regolatorie. Il dominio NBD1 è molto importante per la struttura del canale, tanto che la mutazione più frequente nei malati di fibrosi cistica, la F508del, si trova proprio in questa parte di proteina. Molte ricerche e molti farmaci mirano a stabilizzare NBD1 ma qualche hanno fa è stato dimostrato che esiste un correttore, chiamato VX809, che ripristina CFTR in modo davvero molto efficace e si lega invece ai domini MSD1 e MSD2. Siamo andati a studiare il meccanismo d’azione di VX809 e abbiamo visto che i legame con MSD2 stabilizza in qualche modo anche l’interazione con NBD1 e di conseguenza la funzione di CFTR.
Studiare il meccanismo d’azione di queste molecole è fondamentale per creare combinazioni di farmaci con effetto additivo e sinergico e, così, migliorare la risposta del paziente a un certo trattamento.
Esistono combinazioni di farmaci già approvate per l’uso sui pazienti?
C’è un mix di molecole chiamato Orkambi, già in clinical trial, formato dal correttore VX809 e dal potenziatore VX770. VX770 si lega a MSD2 e stimola l’apertura del canale CFTR, ma stiamo cercando di capire i dettagli di questa interazione per scoprire a quale preciso amminoacido si lega il farmaco. Con questa informazione, i chimici possono modificare la sua struttura molecolare, migliorare l’affinità di VX770 per CFTR e aumentare così la sua efficacia. Il problema di Orkambi, infatti, è che funziona molto bene sulle cellule in vitro ma, una volta passati ai pazienti, la risposta è molto più bassa di quella prevista e c’è una grandissima variabilità da soggetto a soggetto.
A cosa è dovuta questa variabilità?
È quello che stiamo cercando di capire, abbiamo fatto alcune ipotesi ma sono ancora da testare.
Per cercare di ridurla stiamo seguendo un altro approccio, cioè testare correttori e potenziatori su sistemi che siano il più vicini possibile alla condizione in vivo. Abbiamo iniziato a usare le cellule nasali dei pazienti, lavorando in un ospedale pediatrico abbiamo la possibilità di raggiungere moltissimi bambini con fibrosi cistica e attualmente l’obiettivo è di coinvolgerne almeno 100. L’idea è di riuscire a mettere a punto una terapia personalizzata che funzioni nei singoli casi, in modo da diminuire la variabilità della risposta ed evitare di somministrare ai paziente farmaci che non funzionano.
Le cellule nasali vengono prelevate attraverso un metodo poco invasivo, il brushing (spazzolamento) dell’epitelio nasale, poi vengono coltivate in laboratorio e usate sia per testare la risposta alla terapia Orkambi, sia per fare uno screening farmacologico con nuovi composti. Finora ci sono due correttori e un potenziatore che sembrano molto promettenti nel ripristinare il flusso di cloro addirittura a livelli paragonabili ai soggetti sani, cioè senza fibrosi cistica, e persino migliori rispetto all’Orkambi.
Inoltre, questa triplice combinazione si è dimostrata efficace su alcuni pazienti con mutazioni rare. F508del è presente nel 80% dei pazienti, ma esistono più di duemila altre mutazioni a carico di CFTR che meritano comunque attenzione. In Italia, per esempio, una mutazione molto comune è la N1303K, localizzata nel dominio NBD2; purtroppo la triplice combinazioni non mostra effetti positivi su N1303K quindi dovremo ripartire dallo screening farmacologico per selezionare nuove molecole.
Questi screening possono essere fatti anche su cellule provenienti da apparati diversi da quello respiratorio?
In linea di principio sì, ma servirebbero delle biopsie, quindi un intervento invasivo sul paziente, e le cellule ottenute potrebbero essere ricche di batteri, perciò poco utilizzabili. Un’altra strada è partire dalle cellule del sangue, ottenute dai pazienti con un semplice prelievo, e da qui ottenere cellule staminali iPSC (induced Pluripotent Stem Cells) da differenziare in diversi organi. Per ora stiamo cercando di ottenere cellule bronchiali, intestinali ed epatiche, e dai dati preliminari possiamo dire che Orkambi non è efficace a livello intestinale.
Quali sono le prospettive future del tuo lavoro?
Vorremmo iniziare almeno un trial clinico con le molecole che hanno avuto risultati promettenti sulle cellule nasali. Inoltre, stiamo cercando di instaurare una collaborazione con dei gruppi italiani per ottenere cellule nasali con la mutazione N1303K in modo da testare vari farmaci e trovare una combinazione che permetta di ripristinare questo difetto così severo.
Segui Luisa Alessio su Twitter
Leggi anche: Cosa vuol dire avere la fibrosi cistica oggi
Pubblicato con licenza Creative Commons Attribuzione-Non opere derivate 2.5 Italia. ![]()



